|
||
|
|
||
| Главная ≫ Инфотека ≫ Химия ≫ Проблема хиральной чистоты / Биогенез // Михаил Никитин |
Проблема хиральной чистоты / БиогенезМихаил Никитин
Проблема хиральной чистотыНикитин М.А. 
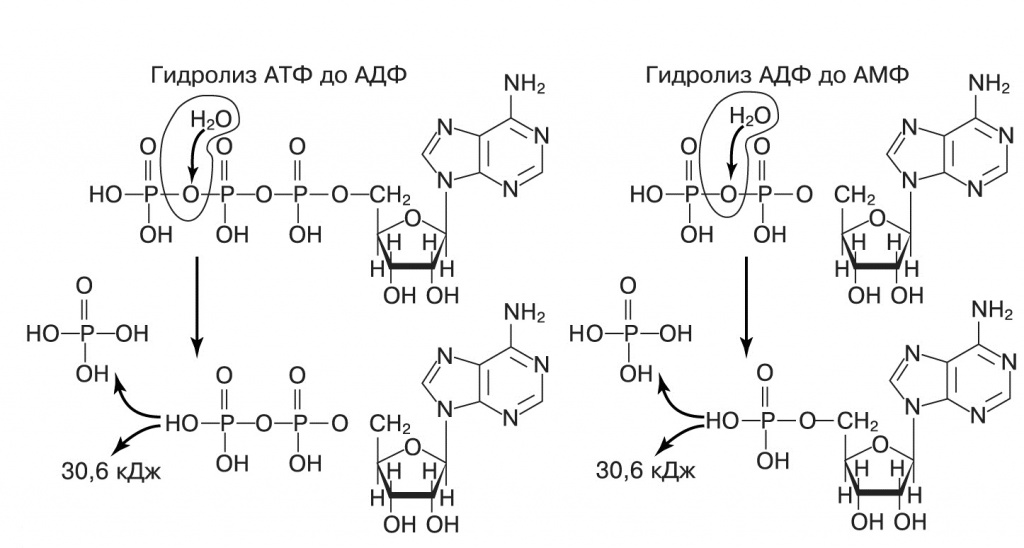
После опытов Миллера, о которых рассказывалось в предыдущем номере, были открыты и другие химические реакции, способные производить органику в условиях древней Земли. Одна из интенсивно изучаемых таких реакций — формозная реакция Бутлерова, открытая еще в 1865 году: водный раствор формальдегида (СH2O) с добавлением Ca(OH)2 или Mg(OH)2 при небольшом нагревании превращается в сложную смесь сахаров. (Об этом, а также о других проблемах биогенеза рассказывалось также в статье В.Н.Пармона «Новое в теории появления жизни», «Химия и жизнь», 2005, № 5.) Формальдегид легко образуется из углекислого газа в присутствии паров воды на поверхности горячего железа — например, на включениях самородного железа, которые содержатся в вулканических лавах при извержениях. Найден он и в кометах, и в межзвездных газовых облаках. Изучению реакции много лет мешал ее капризный характер — колбу с раствором надо было греть несколько часов без всяких видимых изменений, как вдруг за считанные минуты раствор желтел, затем коричневел и загустевал. А если исходные реагенты были очень чистыми, то реакция не шла вовсе. Причиной «капризов» оказался автокаталитический характер реакции: сначала формальдегид медленно превращается в двух- и трехуглеродные сахара (гликоальдегид, глицеральдегид и дигидроксиацетон), которые затем катализируют синтез самих себя и более крупных сахаров. Если к исходной смеси сразу добавить чуть-чуть гликоальдегида или глицеральдегида, то реакция запускается почти сразу. Другой способ ускорить ее — осветить раствор ультрафиолетом, под действием которого отдельные молекулы формальдегида соединяются в гликоальдегид. Обычно в реакции Бутлерова получаются сложные смеси сахаров, где сахара, характерные для живых клеток, перемешаны с огромным разнообразием семи-, восьми-, девятиуглеродных сахаров и даже более сложных. Это долго не давало возможности привлечь ее к предбиогенному синтезу. Однако в последние годы обнаружилось несколько способов, позволяющих избирательно накапливать отдельные сахара, именно те, что нужны для биохимии. Например, при добавлении растворимых силикатов, таких как Na2SiO3, силикат-анион образует комплексы с четырех- и шестиуглеродными сахарами, которые выпадают в осадок и далее не участвуют в реакции. Так накапливаются сахара, имеющие две соседние гидроксильные группы с одной стороны: эритроза, треоза, глюкоза, манноза (подробнее об этом можно прочитать в заметке «Химикам удалось стабилизировать абиогенный синтез сахаров») Если же в реакционную смесь добавить гидроксиапатит Ca3(PO4)2 ∙ Ca(OH)2, то на его поверхности практически избирательно осаждается рибоза (Pestunova et al, «Advances in Space Research», 2005, 36, 214—219, см. также статью В.Н.Пармона в майском номере «Химии и жизни» за 2005 год). Соли борной кислоты тоже избирательно осаждают из реакционной смеси рибозу (Ricardo et al., «Science», 2004, doi:10.1126/science.1092464). Еще один избирательный катализатор реакции Бутлерова — комплекс аминокислоты пролина с ионом цинка. Он также останавливает реакцию на стадии пяти- и шестиуглеродных сахаров, и, что еще важнее, он стереоспецифичен! Комплекс «левого» пролина с цинком избирательно синтезирует «правые» сахара. (Kofoed et al., «Organic and biomolecular chemistry», 2005, doi:10.1039/ b501512j). Ряд других аминокислот, например глутамин и лейцин, тоже обеспечивают стереоспецифичный синтез «правых» сахаров в присутствии «левых» аминокислот, но не останавливают его на стадии рибозы и шестиуглеродных молекул. Откуда же взять «левые» изомеры аминокислот? Как мы уже упоминали, оптические изомеры различаются поведением только при встрече с поляризованным светом или другими оптически активными веществами. Оказывается, в качестве оптически активного партнера могут выступать и другие молекулы того же самого вещества. Вспомним, как Пастер разделил изомеры винной кислоты: при медленном упаривании раствора L- и D-изомеры кристаллизовались отдельно друг от друга, что и позволило рассортировать кристаллы. Если же упаривать раствор с «метеоритным» соотношением изомеров 60:40, то преобладающий изомер начнет выпадать в осадок раньше. Вовремя остановив упаривание, можно получить чистые кристаллы одного изомера и равную их смесь в растворе. Большинство аминокислот ведут себя противоположным образом: при упаривании раствора сначала выпадают рацемические кристаллы (с отношением изомеров 1:1), и раствор обогащается тем изомером, которого было больше в исходной смеси. Так, из раствора фенилаланина с отношением изомеров 52:48 удалось в два цикла упаривания получить раствор с долей L-изомера 90% (Breslow, Levine, «Proceedings of the National Academy of Sciences USA», 2006, doi: 10.1073/pnas.0605863103). Аналогично ведет себя и главный оптически активный промежуточный продукт (и автокатализатор) реакции Бутлерова — глицеральдегид. Пяти- и шестиуглеродные сахара неспособны к такой самоконцентрации оптически активного изомера, но рибоза в составе нуклеозидов (сахар плюс азотистое основание; если присоединить к нуклеозиду остаток фосфорной кислоты, получится нуклеотид) тоже, подобно аминокислотам, предпочтительно кристаллизуется в соотношении изомеров 1:1 и может накапливаться в растворе в оптически чистой форм. (Breslow, Cheng, «Proceedings of the National Academy of Sciences USA», 2010, doi: 10.1073/pnas.1001639107). Более того, в некоторых условиях можно получить хирально чистые аминокислоты из смеси равных количеств обоих изомеров. Группа испанских химиков под руководством Кристобала Видмы («Journal of the American Chemical Society», 2008, doi: 10.1021/ ja8074506) показала, что, если нагреть раствор аспартата в присутствии салицилового альдегида и уксусной кислоты до 100—130ОС, образуются чистые кристаллы одного оптического изомера. Аспартат — это одна из двух аминокислот, оптические изомеры которых кристаллизуются раздельно. Салициловый альдегид в кислой среде катализирует переход изомеров в растворе друг в друга, поэтому небольшие случайные отклонения в начале кристаллизации приводят к полному превращению смеси в чистый L- либо D-изомер. Еще один механизм разделения оптических изомеров — адсорбция на поверхности некоторых минералов. Кристаллы кальцита на одних гранях сильнее удерживают L-аминокислоты, а на других — D-изомеры. Синтез азотистых оснований также может происходить разными путями. Аденин и гуанин образуются из синильной кислоты при замерзании ее водного раствора, ультрафиолетовом облучении или нагревании. Все четыре азотистых основания синтезируются с высоким выходом из формамида NH2CНO на поверхности частиц TiO2 при ультрафиолетовом облучении (Senanayake, Idriss, «Proceedings of the National Academy of Sciences USA», 2006, 103, 1194—1198, doi: 10.1073/pnas.0505768103, Saladino et al., «Chemistry & Biodiversity», 2007, 4, 694—720, doi: 10.1002/cbdv.200790059), аденин, цитозин и урацил — на поверхности монтмориллонита (разновидность глины) или оксидов железа при нагревании (см. обзор Constanzo et al, «BMC Evolutionary Biology», 2007, doi: 10.1186/1471-2148-7-S2-S1). Чтобы азотистые основания приняли участие в синтезе РНК-подобных полимеров, они должны, естественно, сначала объединиться с сахаром и фосфатом. Еще в 1960-е годы было показано, что при ультрафиолетовом облучении раствора аденина, рибозы и фосфатов аденин сначала образует связь с рибозой, а затем присоединяет последовательно три фосфатные группы, превращаясь в АТФ. При этом присоединение последней фосфатной группы происходит примерно в сто раз быстрее, чем предшествующие реакции (Ponnamperuma et al., «Nature», 1963, doi:10.1038/199222a0). Возбужденное триплетное состояние аденина обычно локализует неспаренный электрон на аминогруппе, эта форма легко образует фосфоамидную высокоэнергетическую связь с фосфатом. Далее фосфат переносится на 5'-гидроксильную группу рибозы. Дифосфатная цепь АДФ обладает как раз подходящей длиной для эффективного переноса третьей фосфатной группы. Это, видимо, объясняет, почему в качестве универсального источника энергии в живых организмах используется гидролиз АТФ до АДФ и фосфата, хотя с таким же успехом можно использовать любой нуклеотидтрифосфат и даже дифосфат (его гидролиз до монофосфата выделяет такое же количество энергии). Действительно, ГТФ, ЦТФ, УТФ эпизодически выступают в этой роли, поставляя энергию для некоторых реакций, — однако гидролиз дифосфатов, насколько известно автору, нигде не используется.
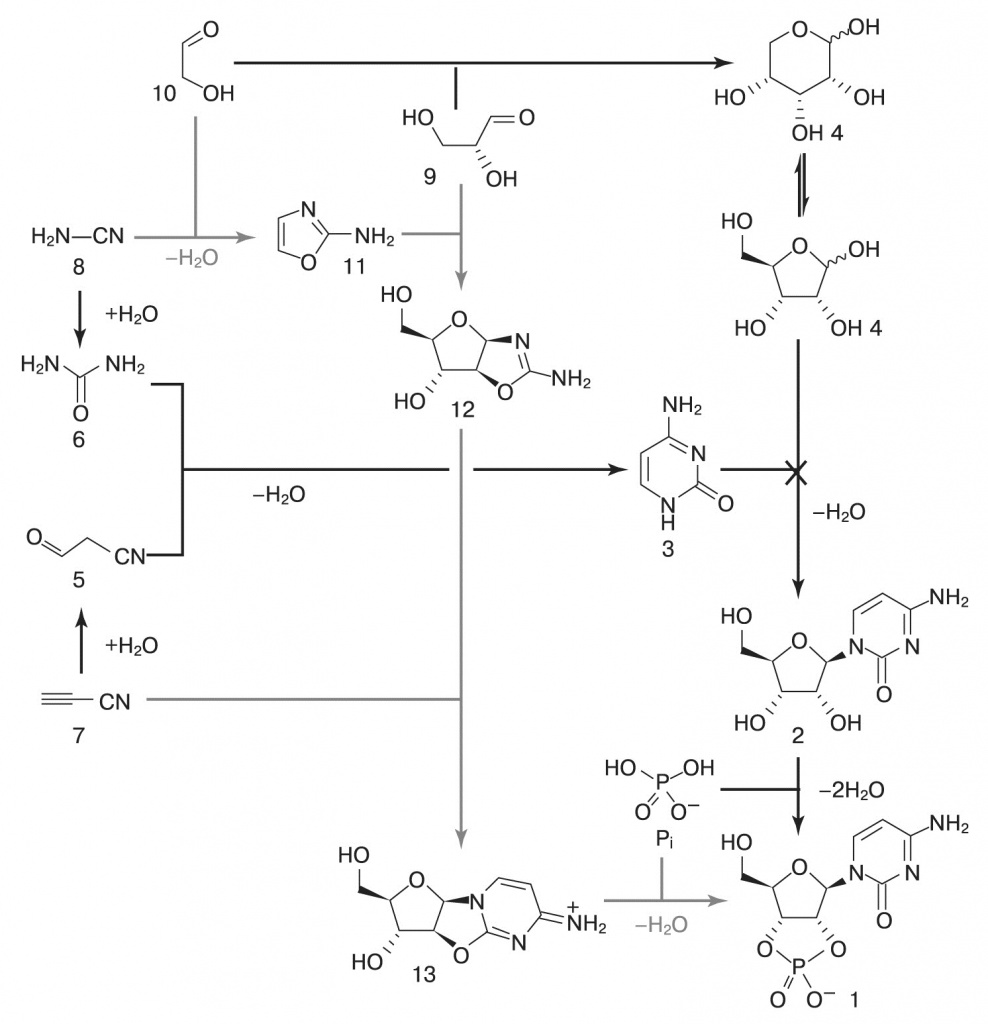
Однако этот способ синтеза активированных нуклеотидов не подходит для других азотистых оснований. Гуанин при облучении присоединяет рибозу, но практически не реагирует с фосфатом. Пиримидины не реагируют и с рибозой. Поэтому очень важной вехой в изучении предбиогенных синтезов стала вышедшая в 2009 году работа Джона Сазерленда с коллегами из Химической школы Манчестерского университета (Powner, Gerland, Sutherland, «Nature», 2009, 459, 239—242, doi:10.1038/nature08013, краткий русский пересказ в заметке «Химики преодолели главное препятствие на пути к абиогенному синтезу РНК»). Они получили активированные пиримидиновые нуклеотиды (циклические 2',3' урацил- и цитидинмонофосфаты), смешивая в одной системе сразу и предшественники сахаров, и предшественники нуклеотидов, и фосфат. Казалось бы, это очень сильно расширяет возможные химические реакции, а значит, побочных продуктов должно быть больше. Однако эксперимент опроверг это предположение.
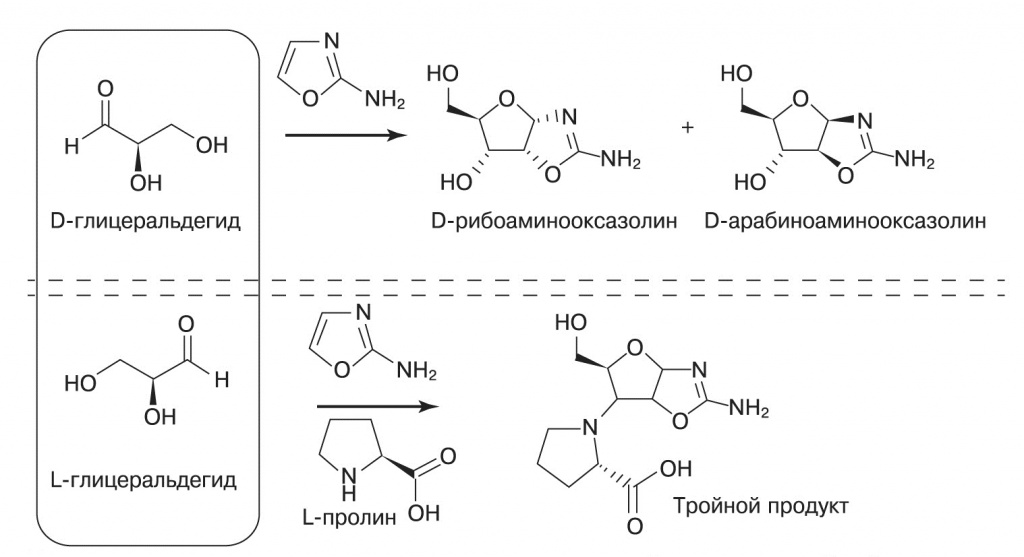
Авторы смешали цианоацетилен (7), цианамид (8), глицеральдегид (9) и гликоальдегид (10). Фосфат избирательно катализирует синтез промежуточных продуктов 11 (2-аминооксазол) и 12 (арабинозоаминооксазолин), подавляя возможные побочные реакции. Затем продукт 12 реагирует с цианоацетиленом, давая вещество 13 (арабинозо- ангидронуклеозид). В обычном водном растворе при этом повышается pH, что приводит к гидролизу промежуточных продуктов и побочным реакциям с цианоацетиленом, но фосфат и тут приходит на помощь, поддерживая среду кислой и направляя реакцию в сторону продукта 13. Для его превращения в циклический цитидинмонофосфат достаточно подогреть реакционную смесь — все необходимое в ней уже имеется. Катали- затором фосфорилирования становится мочевина, образующаяся из цианамида в ходе одной из побочных реакций. На- конец, чтобы избавиться от побочных продуктов этой реакции и превратить часть цитозина в урацил, достаточно ультрафиолетового освещения раствора. Этот синтез поражает своим изяществом: побочные продукты одних реакций здесь становятся катализаторами последующих, фосфат направляет реакции в нужную сторону задолго до того, как войти в окончательный продукт, а ключевой промежуточный продукт (11) способен к самоочищению и накоплению в высоких концентрациях благодаря своей высокой летучести — он хорошо испаряется из водных растворов при слегка повышенной температуре и конденсируется во время ночных заморозков. Как написал редактор журнала «Nature» в предисловии к работе команды Сазерленда: «Именно потому, что эта работа открывает так много новых направлений исследований, она на долгие годы останется одним из великих достижений пребиотической химии». И новые направления исследований немедленно начали развиваться. Уже через два года вышла статья группы Джейсона Хейна (Hein, J.E. et. al., «Nature Chemistry», 2011, 3, 704—706, doi:10.1038/nchem.1108, Hein, J.E., Blackmond, D.G., «Accounts of Chemical Research», 2012, 45, 12, 2045—2054, doi:10.1021/ar200316n). Добавляя к системе Сазерленда различные аминокислоты, они получили стереоспецифический синтез рибонуклеотидов. Более того, достаточно было 1% избытка одного из стереоизомеров аминокислот, чтобы в конце концов получились хирально чистые рибонуклеотиды! Аминокислоты вмешиваются в синтез Сазерленда на стадии реакции 2-аминооксазола с глицеральдегидом, причем образуется тройной продукт. Эта реакция стереоспецифична: пара глицеральдегида с аминокислотой одной хиральности реагирует в четыре раза быстрее, чем разнохиральная. Таким образом, небольшой избыток L-аминокислоты будет связывать L-глицеральдегид в побочный путь реакции, оставляя для синтеза рибонуклеотидов больше D-изомеров.
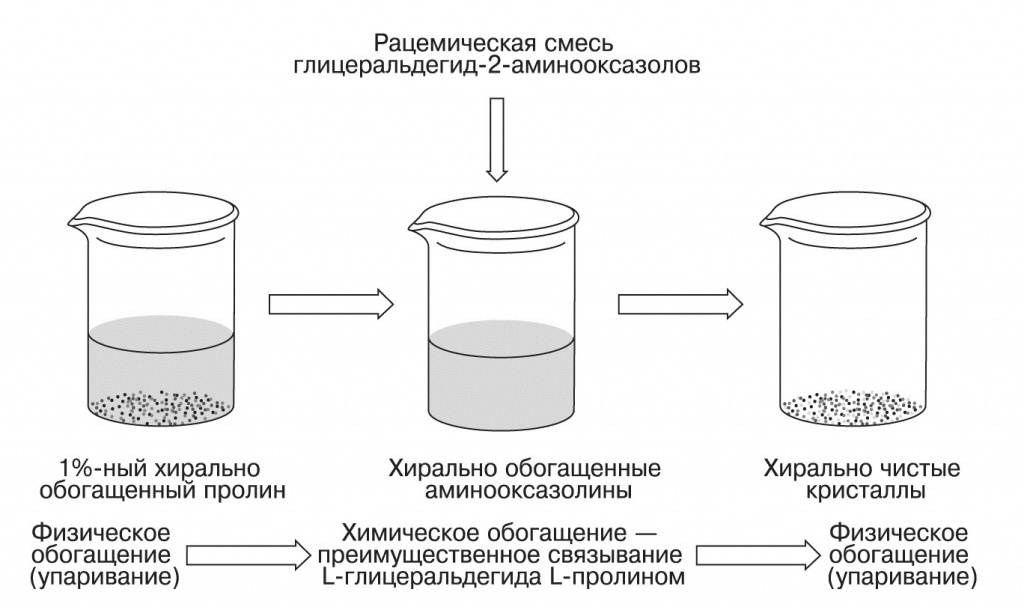
Ранее Сазерленд показал, что рибоаминооксазолин, подобно винной кислоте в опытах Пастера, способен при упаривании раствора кристаллизоваться в хирально чистые кристаллы уже при соотношении изомеров 60:40. Экспериментально получены такие кристаллы рибоаминооксазолина прямо из реакционных смесей с участием 14 чистых L-аминокислот из 19, содержащихся в белках. Пролин по стереоспецифичности далеко превосходит все остальные аминокислоты. Таким образом, достаточно, чтобы в синтез Сазерленда попал раствор аминокислот, хирально обогащенный путем частичной кристаллизации. В экспериментах Хейна так были получены хирально чистые рибонуклеотиды, начиная всего лишь с 1% хирально обогащенного пролина.
Такое небольшое хиральное обогащение аминокислот легко может быть обеспечено фотохимическими процессами с участием поляризованного УФ-света: как мы уже писали, в метеоритах встречаются аминокислоты с хиральным обогащением до 18%, причем с избытком именно L-изомеров. Как видим, проблема разрешима и без вмешательства высшего разума. Литература Guzman, M.I., Martin, S.T. Prebiotic metabolism: production by mineral photoelectrochemistry of alpha-ketocarboxylic acids in the reductive tricarboxylic acid cycle. «Astrobiology», 2009, 9, 833—842, doi: 10.1089/ast.2009.0356. Orgel, L.E. The implausibility of metabolic cycles on the prebiotic Earth. «PLoS Biology», 2008, 6, doi: 10.1371/journal.pbio.0060018. Pasek, M.A. Rethinking early Earth phosphorus geochemistry. «Proceedings of the National Academy of Sciences USA», 2008, 105, 853—858, doi: 10.1073/pnas.0708205105.
ТегиПохожее
|

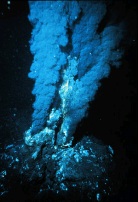 Химики показали, что в гидротермальных источниках при температуре свыше 80 градусов может происходить абиогенный синтез органических веществ, в частности аминокислот, из угарного газа, цианистого водорода и других неорганических соединений. Это открытие — важный аргумент в пользу гипотезы, согласно которой жизнь на Земле зародилась в горячих вулканических источниках.
Химики показали, что в гидротермальных источниках при температуре свыше 80 градусов может происходить абиогенный синтез органических веществ, в частности аминокислот, из угарного газа, цианистого водорода и других неорганических соединений. Это открытие — важный аргумент в пользу гипотезы, согласно которой жизнь на Земле зародилась в горячих вулканических источниках. Эта книга предназначена для широкого круга читателей, желающих узнать больше об окружающем нас мире и о самих себе. Автор, известный ученый и популяризатор науки, с необычайной ясностью и глубиной объясняет устройство Вселенной, тайны квантового мира и генетики, эволюцию жизни и показывает важность математики для познания всей природы и человеческого разума в частности.
Эта книга предназначена для широкого круга читателей, желающих узнать больше об окружающем нас мире и о самих себе. Автор, известный ученый и популяризатор науки, с необычайной ясностью и глубиной объясняет устройство Вселенной, тайны квантового мира и генетики, эволюцию жизни и показывает важность математики для познания всей природы и человеческого разума в частности.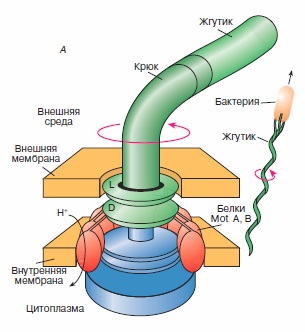 Рассмотрены строение и механизмы действия протонной АТРсинтазы и флагеллярного мотора - молекулярных моторов живой клетки, выполняющих химическую и механическую работу, связанную с их вращательным движением, строение и механизмы работы миозина и кинезина - механохимических белков, ответственных за сократительную активность и внутриклеточный транспорт органелл в клетке.
Рассмотрены строение и механизмы действия протонной АТРсинтазы и флагеллярного мотора - молекулярных моторов живой клетки, выполняющих химическую и механическую работу, связанную с их вращательным движением, строение и механизмы работы миозина и кинезина - механохимических белков, ответственных за сократительную активность и внутриклеточный транспорт органелл в клетке.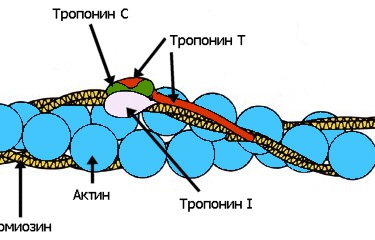 Почти всякая незыблемая общепринятая теория, которую с проклятьями зубрят школьники и которую устало и одинаково рассказывают учителя и даже профессора ВУЗов, при внимательном рассмотрении оказывается отнюдь не однозначной, захватывающей и полной загадок. К теории мышечного сокращения вышесказанное относится в полной мере. В общих чертах она была разработана еще в 50-х годах прошлого века, и классический рисунок с актиновыми и миозиновыми нитями до сих пор кочует из учебника в учебник. Однако реальная картина сокращения мышцы куда запутаннее, интереснее и непонятнее, со множеством подробностей и неожиданных действующих лиц и со сложными ролями, которые исполняют эти лица.
Почти всякая незыблемая общепринятая теория, которую с проклятьями зубрят школьники и которую устало и одинаково рассказывают учителя и даже профессора ВУЗов, при внимательном рассмотрении оказывается отнюдь не однозначной, захватывающей и полной загадок. К теории мышечного сокращения вышесказанное относится в полной мере. В общих чертах она была разработана еще в 50-х годах прошлого века, и классический рисунок с актиновыми и миозиновыми нитями до сих пор кочует из учебника в учебник. Однако реальная картина сокращения мышцы куда запутаннее, интереснее и непонятнее, со множеством подробностей и неожиданных действующих лиц и со сложными ролями, которые исполняют эти лица. Запах корицы и яблок — бабушкин пирог, запах хвои и мандаринов — Новый год, сладкий дурман черемухи — весна… Каждый человек сможет добавить к этому списку длинный ряд своих собственных ассоциаций. Многообразие растительных ароматов, созданных природой, кажется неисчерпаемым, многие из них абсолютно уникальны. Для обозначения таких веществ, которые не принимают непосредственного участия в росте, развитии и репродукции отдельных клеток, более 200 лет назад был предложен термин «вторичные метаболиты». Несмотря на несколько неуважительное название, вещества эти выполняют важную роль в жизни растения в целом, участвуют во взаимодействии растений друг с другом и с окружающей средой. К настоящему моменту идентифицировано более 100000 таких веществ, многие из которых являются легколетучими, и люди воспринимают их как запах растения. В этой лекции я постараюсь рассказать о некоторых особенностях пахучих растений, немного о том, как изучают биосинтез летучих вторичных метаболитов, а также о перспективах применения этих знаний на практике…
Запах корицы и яблок — бабушкин пирог, запах хвои и мандаринов — Новый год, сладкий дурман черемухи — весна… Каждый человек сможет добавить к этому списку длинный ряд своих собственных ассоциаций. Многообразие растительных ароматов, созданных природой, кажется неисчерпаемым, многие из них абсолютно уникальны. Для обозначения таких веществ, которые не принимают непосредственного участия в росте, развитии и репродукции отдельных клеток, более 200 лет назад был предложен термин «вторичные метаболиты». Несмотря на несколько неуважительное название, вещества эти выполняют важную роль в жизни растения в целом, участвуют во взаимодействии растений друг с другом и с окружающей средой. К настоящему моменту идентифицировано более 100000 таких веществ, многие из которых являются легколетучими, и люди воспринимают их как запах растения. В этой лекции я постараюсь рассказать о некоторых особенностях пахучих растений, немного о том, как изучают биосинтез летучих вторичных метаболитов, а также о перспективах применения этих знаний на практике…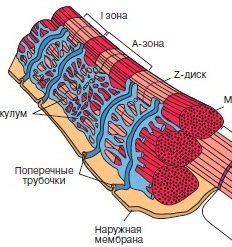 В основе сокращения мышц лежит взаимное перемещение двух систем нитей, образованных актином и миозином. АТФ гидролизуется в активном центре, расположенном в головках миозина. Гидролиз сопровождается изменением ориентации головок миозина и перемещением нитей актина. Регуляция сокращения обеспечивается специальными Са-связывающими белками, расположенными на нитях актина или миозина.
В основе сокращения мышц лежит взаимное перемещение двух систем нитей, образованных актином и миозином. АТФ гидролизуется в активном центре, расположенном в головках миозина. Гидролиз сопровождается изменением ориентации головок миозина и перемещением нитей актина. Регуляция сокращения обеспечивается специальными Са-связывающими белками, расположенными на нитях актина или миозина. Какого цвета могут быть внеземные растения? С научной точки зрения это отнюдь не праздный вопрос, так как цвет поверхности планеты может указать нам, есть ли на ней жизнь, а точнее — живые организмы, усваивающие энергию света своей звезды путем фотосинтеза.
Какого цвета могут быть внеземные растения? С научной точки зрения это отнюдь не праздный вопрос, так как цвет поверхности планеты может указать нам, есть ли на ней жизнь, а точнее — живые организмы, усваивающие энергию света своей звезды путем фотосинтеза. Лекции доктора физико-математических наук, ведущего научного сотрудника сектора математической физики Физического Института им. П.Н. Лебедева РАН, Москва; Directeur de Recherche au CNRS (CNRS — Национальный центр научных исследований) Universite Paris-Sud, Орсэ, Франция Сергея Нечаева, прочитанной 11 апреля 2012 года в рамках проекта «Публичные лекции "Полит.ру"»
Лекции доктора физико-математических наук, ведущего научного сотрудника сектора математической физики Физического Института им. П.Н. Лебедева РАН, Москва; Directeur de Recherche au CNRS (CNRS — Национальный центр научных исследований) Universite Paris-Sud, Орсэ, Франция Сергея Нечаева, прочитанной 11 апреля 2012 года в рамках проекта «Публичные лекции "Полит.ру"»| Главная ≫ Инфотека ≫ Химия ≫ Проблема хиральной чистоты / Биогенез // Михаил Никитин |
|
[time: 10 ms; queries: 7]
4 Июл 2026 00:41:43 GMT+3 |